

Emiliano Brini presents his work on using MELD to predict protein structures and interactions.
Review article on paramagnetic NMR methods is out
Our new paper "Molecular modeling of biomolecules by paramagnetic NMR and computational hybrid methods" is now available from BBA Proteins and Proteomics.
This is intended as an easy to digest review of paramagnetic NMR methods and associated computational modelling strategies.

Lab awarded CIHR grant

I'm extremely proud to announce that the MacCallum lab was awarded a five year grant from CIHR for our project aimed at developing new molecular diagnostics for crystal arthropathies. This is an exciting project with great results to date, and we're grateful for funding to push it further.
Overall success rates in this year's CIHR Project Grant were only 16 percent, which is completely unhealthy and reflects funding that has been flat for 15 years. Success rates outside of BC and Ontario were half the national average. Such regional disparities will not lead to the thriving research environment that Canadians need.
While I'm fortunate to have obtained a grant in the in the current climate, I hope that the Minister of Science recognizes the need for improved funding for science in Canada.
Peter Tieleman awarded Tier 1 Canada Research Chair

We're very happy to report that Peter Tieleman, director of the Centre for Molecular Simulation, was awarded a Tier 1 Canada Research Chair in Molecular Simulation. This award is among the most prestigious in Canada and reflects Peter's fantastic contributions to science.
Visit from Aatto Laaksonen
Professor Aatto Laaksonen visited and gave a CMS seminar on methods for hierarchical coarse graining.

Ken Dill Visits Calgary

It was truly a pleasure to welcome my postdoctoral advisor, Dr. Ken Dill, to visit us in Calgary. Ken's seminar focused on how physical chemistry influences and limits the behaviour of cells. As expected, it was thought provoking, with great engagement from the audience.
MOLSSI Workshop at Stanford

I recently attended the Molecular Sciences Software Institute (MOLSSI) workshop hosted at Stanford. MOLSSI is an NSF-funded consortium that aims to advance molecular simulation software, infrastructure, education, standards, and best practices. The workshop focused on the topics of conformational sampling, molecular simulation workflows, and emerging applications of machine learning in molecular science. The discussion was great and it was a wonderful opportunity to engage with people I hadn't met before, as well as old friends.
Fellowships Awarded

Two postdocs in the lab were recently awarded fellowships.
Congratulations to Kala, who received an AIHS postdoctoral award, and Maggie, who received an NSERC PDF!
Paper featured on F1000
Our recent paper was featured on F1000:
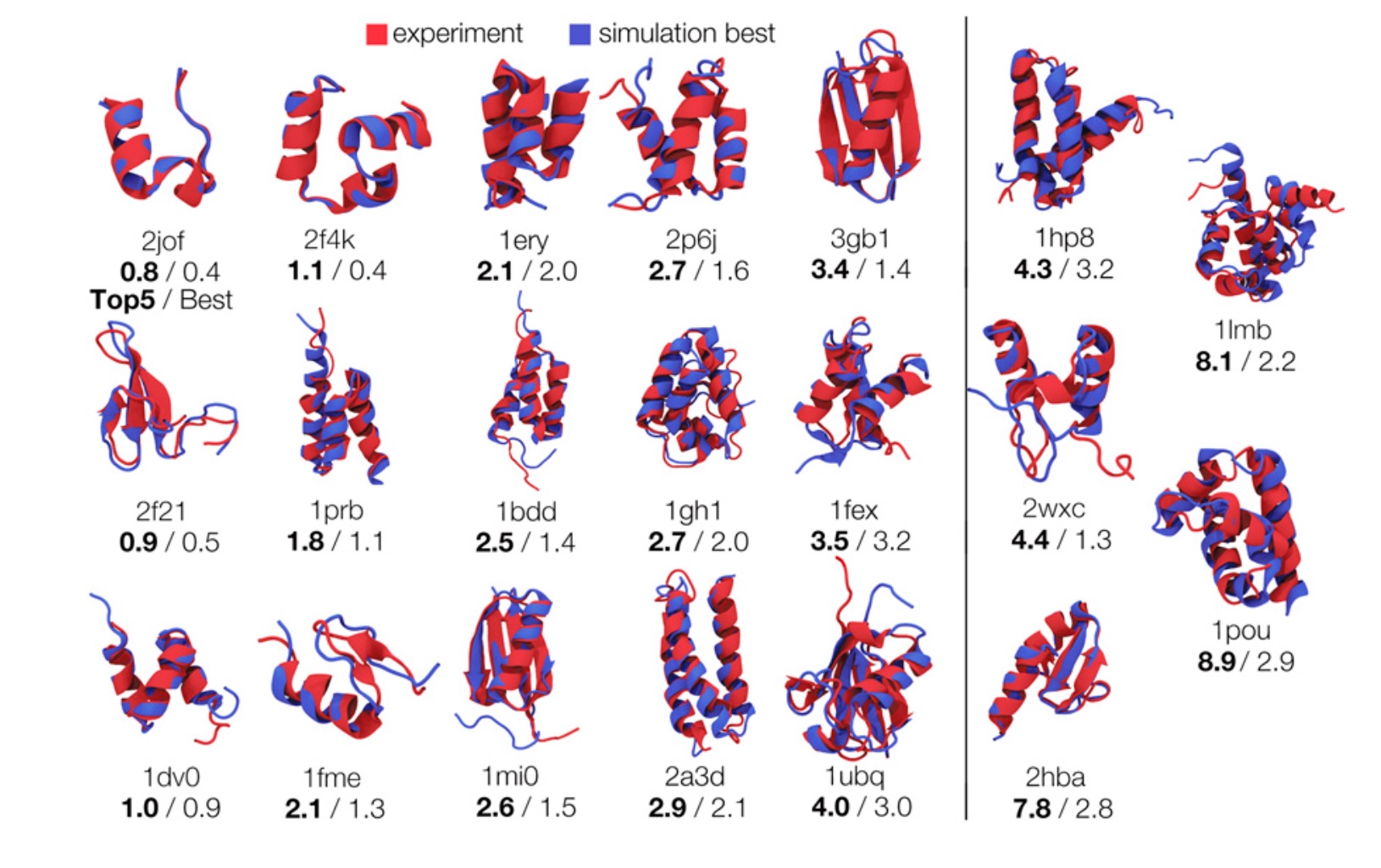
Recent results in the field of protein folding have showed that atomistic molecular dynamics (MD) simulations can correctly fold small proteins, providing insights on their structures as well as on their folding pathways. Those simulations, however, remain computationally expensive. MD simulations are deterministic: once the initial positions and velocities have been set, the trajectory for the protein under study is derived by numerically solving Newton's second law with tiny time steps, hence the long simulation times. If additional information (such as "forming a hydrophobic core") could be added to steer the simulations, it is expected that they could be sped up significantly. However, proper inclusion of such additional information requires caution as it needs to satisfy the thermodynamic principles of Boltzmann's law. The authors have developed a proper statistical mechanics framework for this purpose, they have applied this framework to successfully fold twenty small proteins, including ubiquitin, a millisecond folder. Inclusion of this framework into folding simulations using MD is shown to yield up to five orders of magnitude faster computational folding times than traditional MD.

Protein force field paper published in JCTC
Our latest work on the development of a grid-based correction to the backbone dihedral angles in the Amber forcefield has be published in the Journal of Chemical Theory and Computation.
In this paper, we derive a grid-based correction for use with the Amber ff12SB force field in combination with the GBNeck and GBNeck2 implicit solvation models. This correction improves the relative balance of random coil, alpha helix, and beta sheet secondary structures.

Structure prediction paper published in PNAS
Our latest paper focuses on using weak information to infer protein structures. We use heuristic information—that proteins have hydrophobic cores, secondary structures, and form hydrogen bonds—to accelerate protein folding. The challenge is that we don't know, for example, exactly which residues form hydrogen bonds or how they pair up. However, even this uncertain knowledge is enough to accelerate conformational search by many orders of magnitude.
Adding new forces to OpenMM
Also posted on the omnia.md blog.
I often have colleagues ask why I have switched to OpenMM. As a graduate student I used GROMACS and at the start of my postdoctoral work I used Amber. Both are fine packages. So why switch?
In a word: flexibility. In a few more words: ease of development.
One component of my laboratory's research is integrative structural biology, where we are developing computational methods that can infer protein structures from sparse, ambiguous, and unreliable data. We do this by adding additional forces to the system to account for the experimental data. I won't go into the details of our research, but in this post I will outline the use of custom force classes, which are one of two ways to add custom forces to OpenMM simulations. In a future post, I will discuss the more complex—but also more flexible—route of generating an OpenMM plugin with custom GPU code.
Using custom force classes
The first—and simplest—way to add a new force to OpenMM is to use one of the seven CustomForce classes:
CustomAngleForce
CustomBondForce
CustomExternalForce
CustomGBForce
CustomHbondForce
CustomNonbondedForce
CustomTorsionForce
Each of these classes implements a particular type of interaction. Most of these interaction types are obvious from the name, but a few are worth pointing out. CustomExternalForces represent external forces that act on each atom independent of the positions of other atoms. CustomGBForce allows for the implementation of new types of generalized Born solvation models (see customgbforces.py for examples). Finally, CustomHbondForce is a very interesting class that allows for distance- and angle- dependent hydrogen bonding models—although such terms are not common in standard MD forcefields.
What makes these classes so easy to use is that they do not require writing any GPU code. One simply provides a string containing a series of expressions that describe the force to be added and OpenMM does the rest, differentiating the expression to get the forces and automatically generating the required GPU code.
As an example, the following code implements a flat-bottom harmonic potential.
import openmm as mm
flat_bottom_force = mm.CustomBondForce(
'step(r-r0) * (k/2) * (r-r0)^2')
flat_bottom_force.addPerBondParameter('r0', 0.0)
flat_bottom_force.addPerBondParameter('k', 1.0)
# This line assumes that an openmm System object
# has been created.
# system = ...
system.addForce(flat_bottom_force)Line 3 creates a new CustomBondForce, where the energy to be evaluated is given as a string. It is also possible to use more complex expression with intermediate values (see the manual for more details).
Lines 5 and 6 add per-bond parameters, which are referred to in the energy expression. In this case, for each bond that is added, we will need to specify the values of r0 and k. It is also possible to specify global parameters that are shared by all bonds using the addGlobalParameter method.
Line 11 adds our new force to the system. Don't forget this step! It's easy to overlook. Simply creating the force is not enough, it must actually be added to the system.
Adding Interactions to the system
We've now defined a new custom force. But, how do we add new interactions to our system? Again, OpenMM provides a great deal of flexibility—especially when using the python interface. The code below reads in the flat-bottom restraints to add from a text file.
# restraints.txt consists of one restraint per line
# with the format:
# atom_index_i atom_index_j r0 k
#
# where the indices are zero-based
with open('restraints.txt') as input_file:
for line in input_file:
columns = line.split()
atom_index_i = int(columns[0])
atom_index_j = int(columns[1])
r0 = float(columns[2])
k = float(columns[3])
flat_bottom_force.addBond(
atom_index_i, atom_index_j, [r0, k])Lines 13 and 14 add the new interaction. Notice that the parameters for the bond are specified as a list and that the parameters are given in the order of the calls to addPerBondParameter.
This is a very simple example, but new interactions can be added in a variety of ways: based on atom and residue types, based on proximity to other atoms or residues, based on position in the simulation cell, and so on.
Downsides of the custom force classes
There are only a few potential downsides or pitfalls when using custom force classes. The first is performance. Generally the performance of the custom force classes is quite good. However, as with all automatically generated code, it is likely that a hand-coded version would offer improved performance. However, most interactions will take only a small part of the runtime and spending considerable effort to speed up these interactions is of questionable utility.
The second downside is the limited programability. The custom force classes do not allow for loops, iteration, or if statements. Sometimes one can work around this using functions like min, max, or step (as used above). But sometimes what you want to calculate cannot be easily turned into an expression that the custom force classes use as input. In this case, one must implement a plugin with a hand-coded custom force, which is more flexible, but more involved
Summary
One of OpenMM's key strengths is the ease of adding new types of interactions. I have given a simple demonstration of how to add a custom bonded force. Other types of custom forces like torsions and new non-bonded interactions are similarly straightforward. In a future post, I will outline the steps required to create a custom plugin, which allows for more flexibility and possibly better performance, but at the cost of greater complexity.
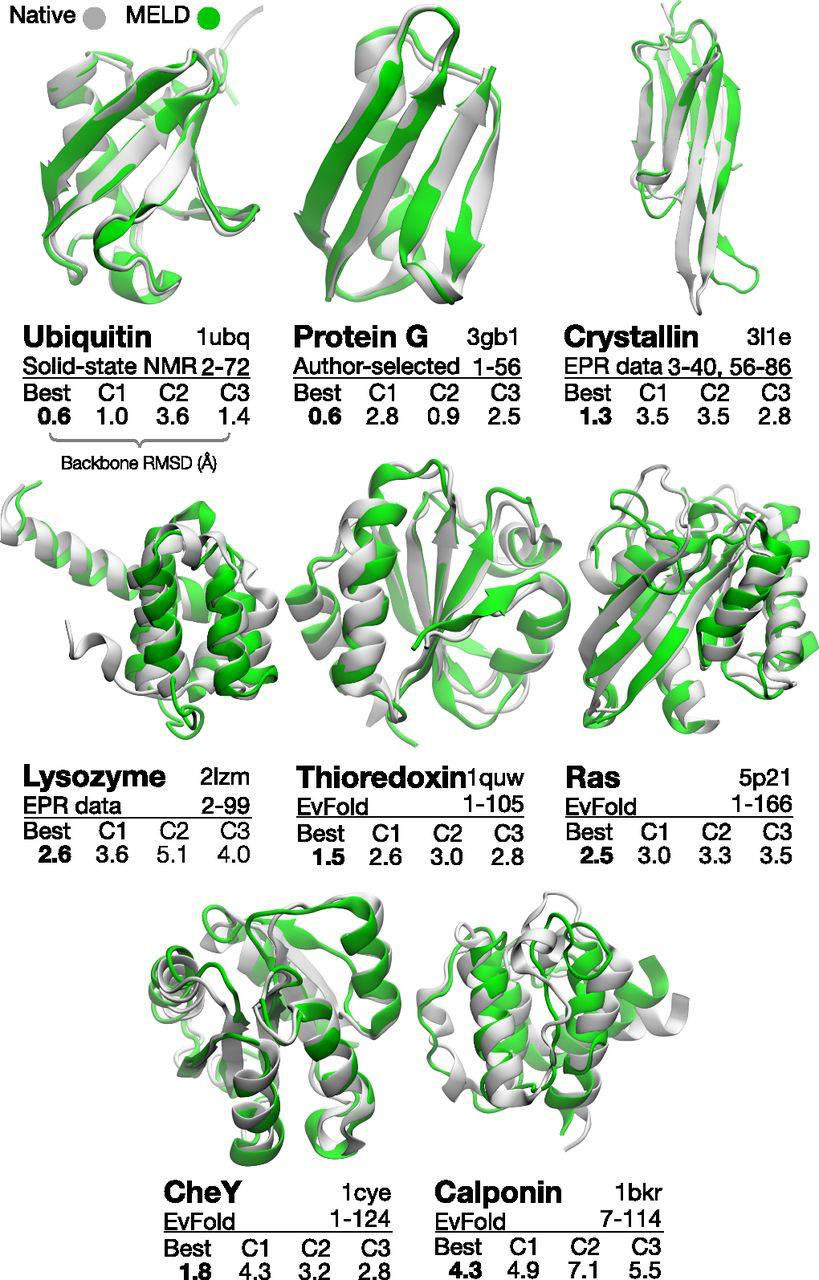
Integrative structural biology paper published in PNAS
My lab's latest paper is now out in PNAS. This work is the first outlining our strategy for integrative structural biology, which allows us to make sense of sparse, ambiguous, and noisy information in structure determination.
Group leader named Canada Research Chair
I'm happy to announce that I've been named as one of the 2015 class of the Canada Research Chairs program.
From the CRC website:
The Canada Research Chairs Program invests approximately $265 million per year to attract and retain some of the world’s most accomplished and promising minds. Chairholders aim to achieve research excellence in engineering and the natural sciences, health sciences, humanities, and social sciences.
In addition to the CRC Chair, I was also awarded $500,000 in funding from Canada Foundation for Innovation, which will help equip my lab with state of the art instrumentation to support our research.
Kananaskis Symposium on Molecular Simulation
We just wrapped up the 6th Kananaskis Symposium on Molecular Simulation.
This three-day retreat is held in beautiful Kananaskis Country at the University of Calgary Biogeoscience Institute's Barrier Lake Field Station.
The retreat featured talks on a variety of simulation-related topics from group leaders, postdocs, and graduate students. There was also plenty of socializing and informal discussion.

This year, David Sivak of Simon Fraser University was our keynote speaker, who gave a fantastic overview of his group's research on non-equilibrium statistical mechanics and its application to biological systems.

30th Annual Darwin Day Celebration
The 30th Annual Darwin Day Celebration at the University of Calgary on Friday, February 6.
I am very excited to see that Dr. Richard Lenski will visiting. Dr. Lenski and co-workers are responsible for the E. coli Long-term Experimental Evolution Project, which has followed the evolutionary fate of several lineages of bacteria for over 60,000 generations. This amazing experiment has been on-going since 1988, and has provided an incredible real-time view of evolution in action.

New shaker arrives
Our new shaker (New Brunswick 44R) arrived today. Tara seems pleased—certainly more so than the crew I assembled to move this beast.
New pipettes have arrived
The Gilson Fairy paid a visit to the lab today and left behind a wall of new pipettes.
Advances in Enhanced Sampling Workshop
This week I'm at the Advances in Enhanced Sampling Workshop in Telluride, CO. The workshop is focused on the latest advances in conformational sampling of biomolecular (and other) systems.
The program has a great line-up of speakers and topics. I'll be giving a talk later in the week on my work on optimizing replica exchange molecular dynamics (REMD) simulations.
Summer School Tutorials Starting
After two days of lectures from our great external speakers, we're now moving into three days of hands-on tutorials. We have three streams:
- Introduction to MD
- Introduction to Martini and coarse-graining
- QM/MM